Harvard Informatics Healthy Habits for Data Science Workshop
Introductions
Good morning and welcome to day 4 of Healthy Habits. Today we're going to talk about how to use scripts and loops to be able to "scale up" analysis, how to deploy these scaled-up analyses on the Cannon cluster, and how to track work with "notebooks". In previous days of the workshop, you've submitted scripts to generate data necessary to produce Figure 1 from our example paper on patterns of expression parallelism of gene expression in E. coli. But today we will take a finer-grained look at SLURM scripts, and explain best practices for quickly and efficiently performing analysis on large numbers of files. In short, we will be explaining, with examples, practical "how tos" for doing with your data, what you've been doing with our example data.
Preliminaries ... workshop data
In case you haven't already done this, please go to the directory you created for running data processing and analysis on the Cannon cluster and do the following:
Running loops in scripts
While in principle, one could manually perform every execution of every step of a pipeline manually--e.g. trimming adaptors from a particular fastq file associated with a particular sample--on the command line, this immediately generates obstacles to efficient analysis, because: * Many analyses can take hours (and some take days) * You don't want to do this in an interactive sessions * As the number of samples increases, becomes very tedious! * Manual execution does not track what you did * Using a generalizable (across samples/datasets) script facilitiates reproducibility
In it's simplest form, executing a command over multiple data sets with a loop works something like this:
for an item in <MY_LIST_OF_ITEMS>
perform <MY_ANALYSIS_COMMAND> on that item
exit the loop after I'm done with the last item
It is worth noting that the list of items need not just be files. They could also be a series of numerical values, if for example you are conducting a simulation and those numbers are values for a specific parameter you are varying. They can also be sub-strings that vary across a bunch of files that only differ by that substring. In general, the "items" can be anything that is required to compose a correct command line with the analysis tool/script being deployed.
Loops and variable assignment in bash: a brief review
The above example of a loop is pseudocode ... but what you need for actual implementation is the real syntax in bash. For example:
seq is just a unix program for generating an iterable array. The above call to seq simply says "create an array that goes from 1 to 10, with a step of 1". Note: you can provide an additional argument to change the step, e.g. seq 1 2 10 would yield: 1,3,5,7,9. So, the above loop says, for eadh element in the array, do something, and in that case that is simply to print to standard output a sentence that ends with $i. The "$" indicates that what follows is a variable, such thatwe are assigning a value for the seq output to the variable i. In short, we assign a value to i, and then we say do the echoing of the string, but in it we include the value of i ... and we distinguish the letter i from the variable i with the "$". There is always a do in a bash for loop, and when the loop is done, we always say "done", i.e. exit the loop.
Can you see where this is going? So, imagine I want to run fastqc --a program to generate various quality control metrics for fastq files-- I might have a series of files: sample1.fq, sample2.fq ... sample10.fq. If I wanted to execute fastqc in a loop:
module load python
source activate fastqc # my fastqc conda environment
for i in seq 1 10
do fastqc --outdir output sample${i}.fq
done
So, we are looping through the values in seq, and then executing fastqc by sticking the variable $i in the command line to select the right sample fastq file. It is often helpful to enclose the variable you are inserting into a command string with {} when it is embedded in a text string (without spaces), to make it unambiguous to the bash interpreter that you are indicating a variable. One could achieve a similar result with the following:
In this case, we are using unix search functionality to list everything that ends with "fq" using the wildcard "*", which means "anything". There will be cases where you need to create an array not simply using a numerical index. We will discuss ways to do this in a little bit when we discuss SLURM job arrays.Cluster architecture as applies to SLURM
SLURM is the scheduler that manages the compute job submissions on Harvard's Cannon cluster. Job submissions invoke the sbatch command:
The SLURM script identifies which specific resources are being requested, and what resources you need to request will depend in part on the nature of the software or code that the job going to run, potentially the size of the data set that the code is going to process, and command line arguments for the code that may affect run time and memory requirements. With this in mind it is important to review some basic high-performance computing (HPC) cluster vocabulary and, specifically, the architecture of Cannon, so that we can relate that architecture to how we request resources.Nodes, CPUs, and cores
What's in a node? CPUs and cores!
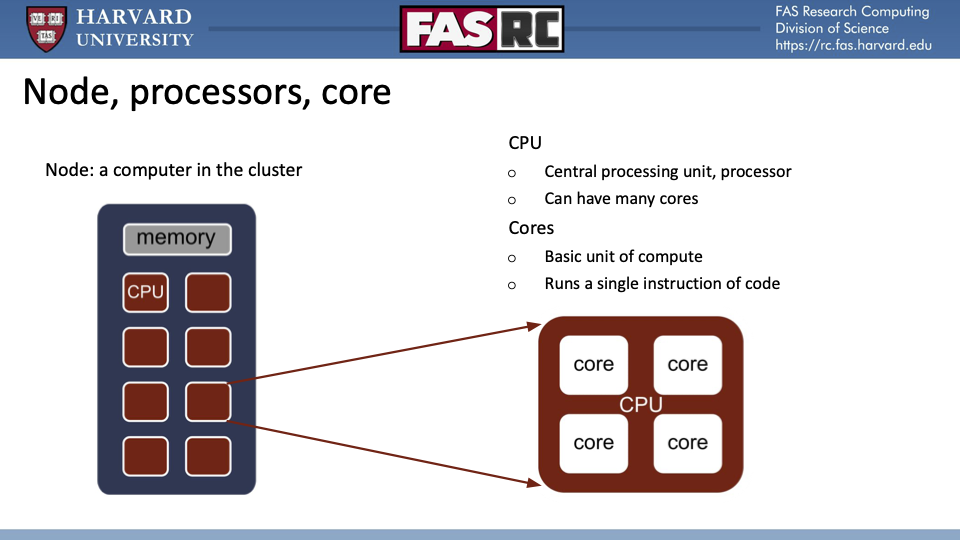
While this distinction between CPUs and cores is meaningful with respect to the actual hardware architecture, from the perspective of end-users of Cannon resources, there is no meaningful distinction. SLURM command line arguments that specify the number of CPUs are actually specifying the number of cores, because they are specifying the indiviual resources that are executing code that gets invoked by command lines. 99%, perhaps more, will fall into one of two categories:
- Jobs that only require the use of a single-core, e.g. running a python script to process a fastq file.
- Jobs that at some point run simultaneously the same analysis across subsets of data, each on a separate core. This type of job is encountered when, for example when runs bwa-mem to align sequencing reads to a genome, by dividing the input fastq file into chunks that are aligned separately.
In both of these cases, the jobs will be run on one node.
A single-core SLURM job script, might look something like this:
#!/bin/sh
#SBATCH --partition=serial_requeue #
#SBATCH -o helloworld.%A.out # stdout log
#SBATCH -e helloworld.%A.err # stderr log
#SBATCH -c 1 # this specifies the number of cores
#SBATCH --mem=2000 # this specifies memory in Mb, i.e. 2Gb in this case
#SBATCH -J # add a short job name here
#SBATCH --time=00:20:00 # this species a job length of 20 minutes
module load python
python myhelloworld.py
A nice trick is using the %A in job logs. This variable stores the SLURM job id, such that if you are running a bunch of jobs in a directory, if when you use sacct to check job status, if a job has failed you can easily use the id to grab the right logs to see what went wrong.
A job that is only using one core, is by definition executing a single task, and a single task will take place on a single node. Running mult-threaded jobs require more careful specification of resources. So, if one were running a bwa-mem job and we want to use 12 threads, we might do something like this
#SBATCH --partition=serial_requeue #
#SBATCH -o bwamem.%A.out # stdout log
#SBATCH -e bwamem.%A.err # stderr log
#SBATCH -n 1 # specifies number of tasks
#SBATCH -N 1 # specifies number of noes
#SBATCH -c 12 # this specifies the number of cores
#SBATCH --mem=24000
#SBATCH -J bwamem
#SBATCH --time=06:20:00 # this species a job length of 20 minutes
bwa mem /BWAMEM/INDEX/PREFIX input_reads_R1.fq input_reads_R2.fq -t $SLURM_CPUS_PER_TASK
There are a few important additions to notice in the multi-threaded case. First, we specify the number of tasks. The bwa-mem job is considered a task ... and internally, it allocates subsets of reads to individual cores. The -c now specifies 12 cores. Because bwa-mem does not run OpenMPI under the hood, it is not capable of running across multiple nodes ... so we require that all requested cores all are on one node. Finally, we can use a convenient SLURM trick, which is to use a SLURM variable $SLURM_CPUS_PER_TASK which equal what is provided to -c. This avoids hard-coding errors, i.e. you want to increase the number of cores, so you change -c but you forget to change a hard-coded value for -t in the command line. SLURM reserves the core resources requested by you--and penalizes your fair share for them--but you only end up using what you provide on the command line.
Data analysis
build kallisto indices with SLURM jobs in a loop
We've thus far learned a little bit about executing loops from the comman line in the shell, and the setup for a SLURM sbatch submission. Let's try putting those two pieces together. To run expression quantification with kallisto, we need to kallisto genome indices by running the index module for kallisto. While, for small bacterial genomes, this could easily and quickly be run on an interactive node, we will practice setting this up in an actual job. As you may recall, there are multiple E. coli genomes that have to be indexed, one for each lineage, so that when we perform quantification on a particular sample, we are doing so with the corresponding index of the correct genome. The "all at once" loop structure would be like this
#!/bin/bash
module load python
conda activate favate
mkdir -p alignment/kallisto/indices
for file in fastas/*; do
# derive an index name from the file name
name=`echo $file | cut -d '/' -f 4 | cut -d '.' -f 1`_k19.kidx
# create the index
kallisto index -i alignment/kallisto/indices/$name $file -k 19 &
done
Exercise:
- Copy kallisto_index.sh that you grabbed with cp at the start of today's workshop, and rename that copy kallisto_index_noloop.sh
- Add a SLURM header so that the script runs for 5 minutes with 1Gb of memory on serial requeue
- Change log files and job name accordingly, and have log files write into a directory called index_logs.
- You will then need to modify the code that it is no longer a loop, but takes a command line argument of a single fasta file.
- In other words, we want a script that executes one kallisto index command per job. HINT: we will use a loop to iterate over the genome fastas so we can supply the fasta name as a command line argument.
- You should also remove the mkdir line from the code and just execute that on the command line so that you aren't trying to create the directory holding the index directories over and over again
- Change the command that assigns a value to name in the loop above so that the cut command on '/' is -f 2 NOT -f 4. Otherwise, you will be grabbing a field from cut that doesnt exist!
- Finally, use your new script to submit your kallisto indexing jobs. The syntax of the submission should be as follows:
Introducing ... job arrays!
With a limited number of input files, or variations on a command line argument you are permuting to investigate its effects on output, it may be straightforward to feed input arguments in a loop (as above). As the number of files or inputs increases, this becomes a less ideal way of doing things. A (frequently better) alternative is to submit a set of related jobs that use the same general command structure, as a job array. In a SLURM job array there is a parent job (with its own id) and a series of associated child jobs subsumed by it: one for each command line execution with the specific inputs you have provided. If you check the job status of an array with sacct, it will return something like this:
| JobID | JobName | Partition | Account | AllocCPUS | State | ExitCode |
|---|---|---|---|---|---|---|
| test | serial_requeue | 1 | RUNNING | 0:0 | ||
| test | serial_requeue | 1 | RUNNING | 0:0 | ||
| . | . | . | . | . | . | . |
| . | . | . | . | . | . | . |
| . | . | . | . | . | . | . |
| test | serial_requeue | 1 | RUNNING | 0:0 |
where the 1,2 .... 1000 represent the value for the $SLURM_ARRAY_TASK_ID, i.e. the ids of those child jobs. If you run sacct on the parent job, you will get the status of all the child jobs. And, conveniently, if you want to see if any of the child jobs failed, you can do something like this:
sacct -j $jobid --format=JobID%20,State|grep -v COMP |grep -v "batch\|extern" |awk -F"\t" '{print $1,$3}'
So how do we build job arrays in SLURM? Well ... there are two ways.
1st way: naming inputs to contain $SLURM_ARRAY_TASK_ID
Imagine you have generate a set of predicted CDS transcripts as part of a genome annotation pipeline for a new genome assembly, and you want to see if they have BLAST hits to real sequences, specifically to the Uniref90 database. You've downloaded the database and built a BLAST index for it. Let's say you have 50000 predicted CDS transcripts in it. Running BLASTX to Uniref90 as one job would take a long time, so you split your newassembly_CDS.fa into 100 fastas with approximately 500 sequences in each, such that they are labelled newassembly_CDS_1.fa, newassembly_CDS_2.fa ... newassembly_CDS_500.fa. An easy way to run a job array for the BLASTX searches is to equate the number strings in the file names asto values for the $SLURM_ARRAY_TASK_ID variable. Then, your script would look something like this:
#!/bin/bash
#SBATCH -J blastx
#SBATCH -N 1
#SBATCH -n 8
#SBATCH -p serial_requeue,shared # Partition
#SBATCH -t 16:00:00
#SBATCH --array=1-500
#SBATCH --mem=8000
#SBATCH -o logs/blstx_%A_%a.out
#SBATCH -e logs/blstx_%A_%a.err
module load python
source activate blast_2.12.0
# below, assuming that all the blast database files have Uniref90.faa as their prefix
blastx -max_target_seqs 5 -num_threads 8 -evalue 1e-5 -query newassembly_CDS_${SLURM_ARRAY_TASK_ID}.fasta -outfmt 6 -db /PATH/TO/Uniref90.faa > newassembly_CDS_blastx2uniref90.${SLURM_ARRAY_TASK_ID}.tsv
In the above script, the log files contain two SLURM variables. We have already seen %A for non-array jobs and in this case it represents the parent jobID. For array jobs, the %a% is the child job id, i.e. the value for \$SLURM_TASK_ID that is assigned to that specific fasta file fed in as input. Besides using \$SLURM_ARRAY_TASK_ID to specify the input file, we can (and should) also embed it in the output file. Once all the array child jobs are done we can concatenate the results into one large file.
2nd way: combining bash arrays with SLURM job arrays
In many cases, you may want to iterate over a set of files that don't have a conveniently embedded numeric string that you can equate with $SLURM_ARRAY_TASK_ID. In cases such as these, you want to: * Create a bash array for the files, then use the $SLURM_ARRAY_TASK_ID as an index to grab a specific value in the array.
Bash arrays are in many ways similar to python lists (or arrays), and the positions in the bash array, like python, start with zero. Suppose that, instead of running inividual kallisto index as a series of discrete SLURM jobs, we wanted to run indexing as an array job...how would we do that? First, we would want to create the bash array. If we look at the contents of the fastas directory:
[afreedman@holylogin02 healthy-habits]$ ls *fastas
AraM1.fasta AraM3.fasta AraM5.fasta AraP1.fasta AraP3.fasta AraP5.fasta AraR6.fasta ecoli_rrna.fasta
AraM2.fasta AraM4.fasta AraM6.fasta AraP2.fasta AraP4.fasta AraP6.fasta AraR7.fasta
This calls the readarray software, and sticks the list of files in an array called files. To list the first entry of the array, one does as follows:
To list all of the entries, one uses the "@" character:
echo ${files[@]}
fastas/AraM1.fasta fastas/AraM2.fasta fastas/AraM3.fasta fastas/AraM4.fasta fastas/AraM5.fasta fastas/AraM6.fasta fastas/AraP1.fasta fastas/AraP2.fasta fastas/AraP3.fasta fastas/AraP4.fasta fastas/AraP5.fasta fastas/AraP6.fasta fastas/AraR6.fasta fastas/AraR7.fasta fastas/ecoli_rrna.fasta
#!/bin/bash
#SBATCH --partition=serial_requeue
#SBATCH -o kalindex_%A_%a.out
#SBATCH -e kalindex_%A_a.err
#SBATCH -n 1
#SBATCH --mem=1000
#SBATCH -J kalindex
#SBATCH --time=00:15:00
readarray -t files < <(ls fastas/*fasta)
file=${files[${SLURM_ARRAY_TASK_ID}-1]}
echo "file is $file"
name=`echo $file | cut -d '/' -f 2 | cut -d '.' -f 1`_k19.kidx
echo "index name is $name"
kallisto index -i alignment/kallisto/indices/$name $file -k 19
We are not going to submit this array job as we've already created the kallisto indices.
kallisto quant job array
Now, we will use a pre-existing sbatch script so that we can run a kallisto quant job array to get expression estimates for our samples. At this point, your directory structure within your day4/ directory should look like this
and the contents of alignment/kallisto/ should look like this:
and finally, the contents of data/ should look like this:
ls data/6-rrna_depleted/
rep1-rna-AraM1.fq.gz rep1-rna-AraM5.fq.gz rep1-rna-AraP3.fq.gz rep1-rna-AraR6.fq.gz rep2-rna-AraM3.fq.gz rep2-rna-AraP1.fq.gz rep2-rna-AraP5.fq.gz
rep1-rna-AraM2.fq.gz rep1-rna-AraM6.fq.gz rep1-rna-AraP4.fq.gz rep1-rna-AraR7.fq.gz rep2-rna-AraM4.fq.gz rep2-rna-AraP2.fq.gz rep2-rna-AraP6.fq.gz
rep1-rna-AraM3.fq.gz rep1-rna-AraP1.fq.gz rep1-rna-AraP5.fq.gz rep2-rna-AraM1.fq.gz rep2-rna-AraM5.fq.gz rep2-rna-AraP3.fq.gz rep2-rna-AraR6.fq.gz
rep1-rna-AraM4.fq.gz rep1-rna-AraP2.fq.gz rep1-rna-AraP6.fq.gz rep2-rna-AraM2.fq.gz rep2-rna-AraM6.fq.gz rep2-rna-AraP4.fq.gz rep2-rna-AraR7.fq.gz
In other words, there are kallisto indices for the E. coli lineages in alignment/kallisto/indices, and there are the cleaned-up rRNA-depleted RNA-seq fastq files in data/6-rrna_depleted.
The script we will use to run kallisto quant looks like this:
#!/bin/bash
#SBATCH -J kallquant
#SBATCH -N 1
#SBATCH -n 4
#SBATCH -t 8:00:00
#SBATCH -p serial_requeue,shared
#SBATCH --mem=24000
#SBATCH -o quant_logs/kallisto_quant_%A_%a.out
#SBATCH -e quant_logs/kallisto_quant_%A_%a.err
#SBATCH --array=1-28
##SBATCH --mail-type=ALL
module purge
module load python
conda activate favate
readarray -t files < <(ls data/6-rrna_depleted/*.fq.gz)
file=${files[${SLURM_ARRAY_TASK_ID}-1]}
echo "file is $file"
lineage=`basename $file |awk -F"-" '{print $3}' | sed 's/.fq.gz//g'`
echo "lineage is $lineage"
library=`basename $file |sed 's/.fq.gz//g'`
echo "library is $library"
index=alignment/kallisto/indices/${lineage}_k19.kidx
echo "index is $index"
mkdir -p alignment/kallisto/output
output=alignment/kallisto/output/${library}_quant
echo "output is $output"
kallisto quant -i $index -o $output --single -l 25 -s 6 -t 4 $file
It should be run from the day4/ directory as follows:
It has to be run from there so that relative paths to the kallisto indices and the fastq files are correct: these paths are relative to where the sbatch command is executed ... NOT where the actual script resides. Also, because the fastq files are read in as an array from their location, with sample names parsed from the files, an necessary directories created accordingly, one does not have to supply any command line arguments.
The job efficency interlude ...
How long it takes for your cluster job to get running depends upon your position in the job queue. That position is determined by your "fairshare" and more specifically, that of your lab. If you and your lab members launch a ton of large-resource requiring jobs (lots of memory, lots of nodes), it will reduce your fairshare and likely increase your wait time befor jobs switch from PENDING to RUNNING status. Thus .. it is worth giving some thought to how you choose resources. For more about how fairshare is calculated, check out FASRC Fairshare and Job Accounting. There are a few key things to consider when requesting resources in a SLURM job:
- Your requested number of nodes,cores and memory are held for you during the entire length of the job, whether your job uses them or not! The reduction in fairshare will get determined by the resources *during the time the job was running.
- In other words, your fairshare is not affected by the time after your job completes, but before you reach the time limit in your job settings. However, requesting excessively long run times will affect how long it takes for your job to start, because the scheduler has to find the node(s) that are available for that long.
- If you run jobs that request unneeded large amounts of compute resource, it will affect your lab fairshare ... and make your lab mates rather unhappy with you.
This is why ... it is worth running a few initial jobs to check how efficient your resource request is. This is why ... seff is your friend. Seff is a bit of code that comes with SLURM that can tell you about your resource use.
So, let's look at my kallisto quant job's efficiency. We can run seff on the array parent job, summarizing across all child jobs, or just do individual jobs:
seff 24865584_1
Job ID: 24865585
Array Job ID: 24865584_1
Cluster: odyssey
User/Group: afreedman/informatics
State: COMPLETED (exit code 0)
Nodes: 1
Cores per node: 4
CPU Utilized: 00:00:16
CPU Efficiency: 25.00% of 00:01:04 core-walltime
Job Wall-clock time: 00:00:16
Memory Utilized: 147.83 MB
Memory Efficiency: 0.62% of 23.44 GB
seff 24865584_12
Job ID: 24865596
Array Job ID: 24865584_12
Cluster: odyssey
User/Group: afreedman/informatics
State: COMPLETED (exit code 0)
Nodes: 1
Cores per node: 4
CPU Utilized: 00:00:13
CPU Efficiency: 54.17% of 00:00:24 core-walltime
Job Wall-clock time: 00:00:06
Memory Utilized: 227.76 MB
Memory Efficiency: 0.95% of 23.44 GB
seff 24865584
Job ID: 24865584
Array Job ID: 24865584_28
Cluster: odyssey
User/Group: afreedman/informatics
State: COMPLETED (exit code 0)
Nodes: 1
Cores per node: 4
CPU Utilized: 00:00:08
CPU Efficiency: 50.00% of 00:00:16 core-walltime
Job Wall-clock time: 00:00:04
Memory Utilized: 181.88 MB
Memory Efficiency: 0.76% of 23.44 GB
#!/bin/bash
#SBATCH -J kallquant
#SBATCH -N 1
#SBATCH -n 4
#SBATCH -t 8:00:00
#SBATCH -p serial_requeue,shared
#SBATCH --mem=24000
#SBATCH -o quant_logs/kallisto_quant_%A_%a.out
#SBATCH -e quant_logs/kallisto_quant_%A_%a.err
#SBATCH --array=1-28
##SBATCH --mail-type=ALL
Jobs that requested 8 hours per array execution took a few seconds ... and certainly DID NOT use 24Gb! Overall ... it seems pretty clear that we could request far less memory, fewer cores, and far less time to run the jobs!